

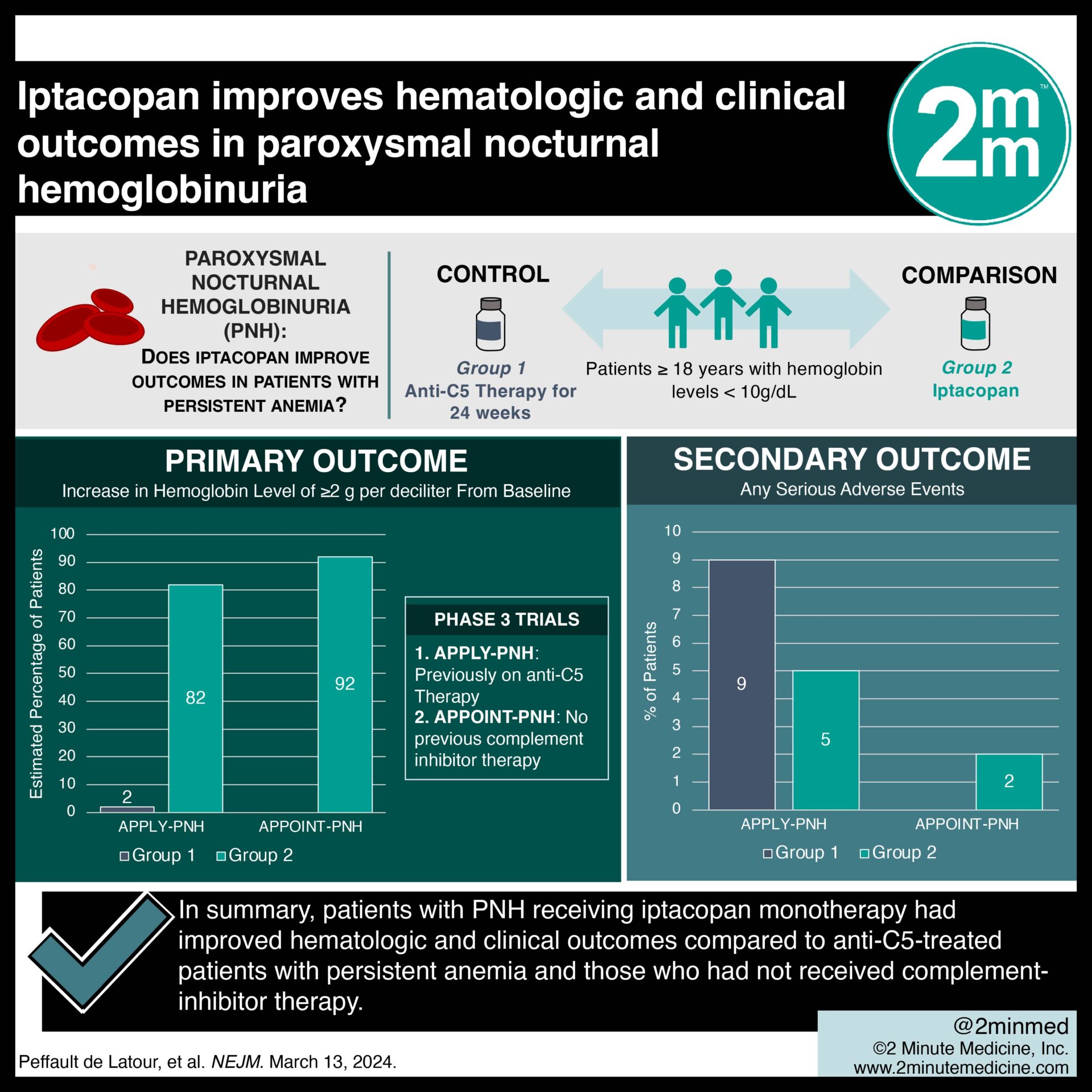
 1. In two clinical trials, iptacopan in patients with paroxysmal nocturnal hemoglobinuria (PNH) improved hemoglobin levels compared to anti-C5-treated patients and those without complement inhibitors.
1. In two clinical trials, iptacopan in patients with paroxysmal nocturnal hemoglobinuria (PNH) improved hemoglobin levels compared to anti-C5-treated patients and those without complement inhibitors.
2. Patients who received iptacopan generally exhibited improved secondary outcomes, including less fatigue, reduced reticulocyte and bilirubin levels, and decreased breakthrough hemolysis.
Evidence Rating Level: 1 (Excellent)
Study Rundown: PNH is a rare, acquired disease characterized by complement-mediated hemolysis, thrombosis, and bone marrow failure. A mutation in the PIGA gene (phosphatidylinositol N-acetyl glucosaminyltransferase, subunit A) renders affected red blood cells deficient in complement regulators CD55 and CD59 and, in turn, more susceptible to intravascular hemolysis. Eculizumab, an anti-C5 monoclonal antibody, and ravulizumab, its long-acting derivative, became standard treatment due to their intravascular hemolysis control, reduced thromboembolic risk, and improved long-term survival. However, some patients with PNH continue to exhibit anemia despite anti-C5 therapy due to extravascular hemolysis from proximal complement pathways. Iptacopan, an oral factor B inhibitor targeting the alternative pathway, has demonstrated an ability to maintain intravascular and extravascular hemolysis control in PNH patients with persistent anemia despite anti-C5 therapy in several phase II trials. In two phase III trials, the effects of iptacopan monotherapy on the hemoglobin levels of PNH patients were investigated. Overall, the trials demonstrated that patients who received iptacopan for 24 weeks exhibited improved hemoglobin levels compared to those who received anti-C5 therapy and those who had not received complement inhibitors. Both trials had a relatively short follow-up period of 24 weeks, limiting their ability to assess the risk of complement-amplifying adverse events such as thromboembolism and infection.
Click to read the study in NEJM
In-Depth [randomized controlled trial]: APPLY-PNH, a randomized controlled trial involving PNH patients with persistent anemia despite anti-C5 therapy, and APPOINT-PNH, a single-group trial involving PNH patients who had not received complement-inhibitor therapy, are two phase III, open-label trials which investigated iptacopan monotherapy for the treatment of PNH. Both trials enrolled patients 18 or older with hemoglobin levels less than 10 g/dL. In APPLY-PNH, patients were randomly assigned in an 8:5 ratio to receive iptacopan or continue anti-C5 therapy for 24 weeks. The primary endpoints of APPLY-PNH included an increase in baseline hemoglobin level by at least 2 g/dL and a hemoglobin level of at least 12 g/dL without red-cell transfusion. The primary endpoint of APPOINT-PNH was an increase in baseline hemoglobin level by at least 2 g/dL. In APPLY-PNH, 51 of 60 evaluable patients in the iptacopan group had a hemoglobin increase of at least 2 g/dL (estimated percentage, 85%; 95% confidence interval [CI], 73 to 90) compared to 0 of 35 patients in the anti-C5 therapy group (estimated percentage, 2%; 95% CI, 1 to 4) (difference, 80 percentage points; 95% CI, 71 to 88; p<0.001). An estimated 69% of patients in the iptacopan group (95% CI, 58 to 79) and 2% of patients in the anti-C5 group (95% CI, 1 to 4) reached a hemoglobin level of at least 12 g/dL (difference, 67 percentage points; 95% CI, 56 to 77; P<0.001). In APPOINT-PNH, 31 of 33 evaluable patients (estimated percentage, 92%; 95% CI, 82 to 100) had a hemoglobin increase of at least 2 g/dL. Serious adverse events occurred in 10% and 14% of patients in the iptacopan and anti-C5 groups, respectively, in APPLY-PNH. In summary, patients with PNH receiving iptacopan monotherapy had improved hematologic and clinical outcomes compared to anti-C5-treated patients with persistent anemia and those who had not received complement-inhibitor therapy.

















